Comment sont réalisés les essais cliniques
Les acteurs des essais
Un essai clinique implique la participation d’un grand nombre de personnes, chacune ayant un rôle bien précis :
- Le promoteur : ce peut être une personne physique, mais il s’agit généralement d’une institution de recherche (une association de médecins, un institut de recherche par exemple) ou d’un laboratoire pharmaceutique. C’est le promoteur qui décide de lancer l’essai, qui assure son organisation et son financement, et qui en est responsable auprès des autorités de santé ;
- L’investigateur principal : c’est le médecin qui coordonne la réalisation de l’essai, de l’élaboration du protocole jusqu’à l’analyse et la publication des résultats ;
- Les investigateurs : ce sont des médecins qui assurent la prise en charge des malades inclus dans l’essai selon les modalités définies dans le protocole.
- Les attachés de recherche clinique (ARC) : ils ont pour mission la mise en œuvre des essais dans les hôpitaux et d’en contrôler la bonne réalisation sur le plan scientifique, technique et réglementaire.
- Les techniciens d’étude clinique (TEC) : ils sont chargés notamment du recueil et de la saisie des données prévues par le protocole des essais.
- Les statisticiens : ils ont notamment pour objectif de déterminer, à partir de la question posée par un essai et de différentes hypothèses statistiques, le nombre de patients qui doivent participer à ce dernier.
- L’Agence nationale de sécurité des médicaments et des produits de santé (ANSM) : c’est un établissement public placé sous la tutelle du ministère de la Santé. Avant de pouvoir débuter, tout essai doit être approuvé par l’ANSM. L’agence s’assure notamment que la sécurité des patients participant à l’essai sera garantie.
- Les Comités de protection des personnes (CPP) : agréés par le ministère de la Santé, les CCP sont composés à parts égales de professionnels de santé et de représentants de la société civile. Ces comités agissent en toute indépendance. Le protocole de tout essai doit obtenir l’avis favorable d’un CPP. Ce dernier s’assure notamment que les droits des malades qui vont y participer sont bien respectés.
- Les malades : les malades qui participent à un essai clinique sont des acteurs à part entière de ce dernier.
Les différentes phases des essais cliniques
Pour évaluer de la façon la plus complète possible un nouveau médicament, un nouveau traitement ou un nouveau mode de prise en charge, différents types d’essais cliniques sont successivement réalisés. Chaque type correspond à ce que l’on appelle une phase.
- Les essais de phase I. Ils sont également appelés « essais précoces ». Les essais de phase I ont pour but d’évaluer la première administration à l’homme d’un nouveau médicament. Ils sont généralement réalisés avec la participation d’un nombre réduit de malades (de 20 à 40 en général). Ce sont toujours des patients qui participent aux essais de phase I en cancérologie, alors que dans d’autres domaines médicaux, il s’agit généralement de « volontaires sains », c’est-à-dire de personnes en bonne santé. Ce type d’essai vise notamment à déterminer la dose maximale tolérée, c’est-à-dire la dose de médicament au-delà de laquelle les effets indésirables deviennent trop importants. Ils permettent ainsi de déterminer la dose qui sera ensuite utilisée dans les essais ultérieurs. Les essais de phase I permettent également d’obtenir des premières informations sur la façon dont le médicament se diffuse dans l’organisme (ce que l’on appelle la pharmacocinétique).
- Les essais de phase II. Ce type d’essai sert à déterminer de façon définitive la dose du médicament associée à la meilleure efficacité et à la meilleure tolérance possible. Ils apportent des informations plus précises sur la diffusion du médicament dans l’organisme et des premières données d’efficacité. Les essais de phase II sont généralement réalisés avec un nombre limité de malades (plusieurs dizaines).
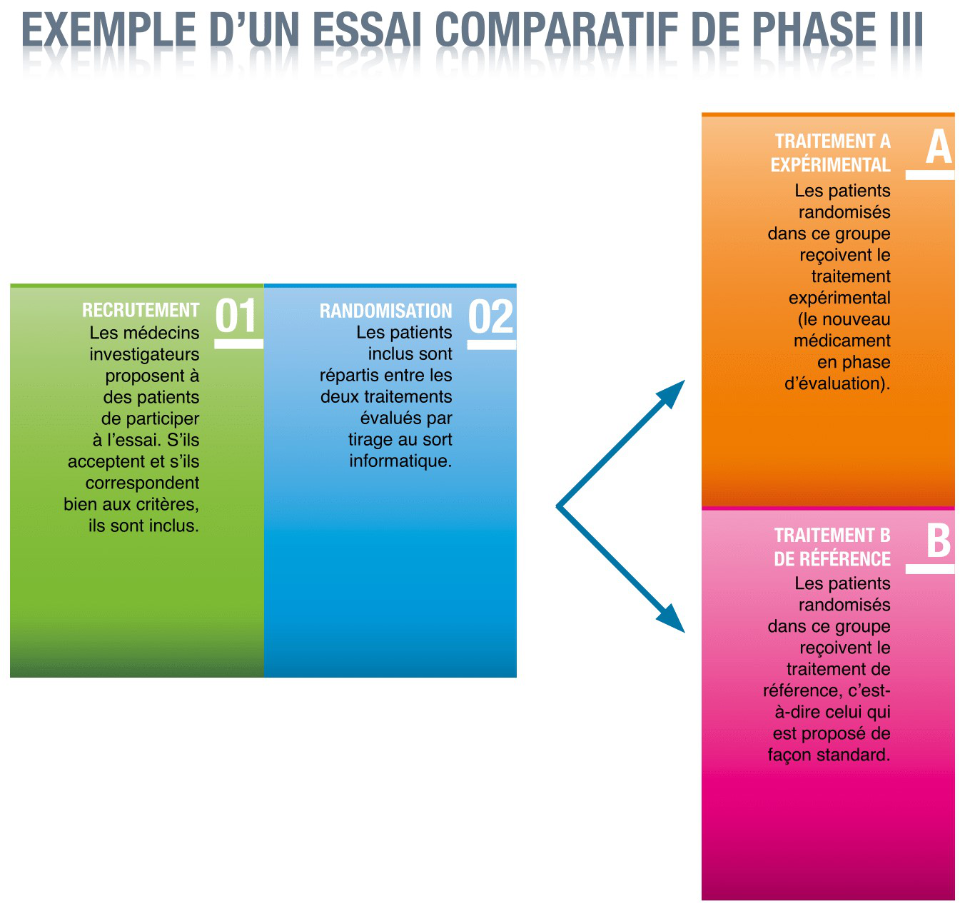
- Les essais de phase III. Ce sont les essais cliniques qui permettent d’évaluer l’efficacité d’un médicament, d’un traitement ou d’un mode de prise en charge. Ils sont toujours comparatifs, c’est-à-dire qu’ils comparent un nouveau médicament (ou nouveau traitement) à un médicament (ou traitement) dont l’efficacité est déjà bien établie et qui fait ainsi office de référence. Ces essais nécessitent la participation d’un grand nombre de malades (souvent plusieurs centaines, voire plusieurs milliers). Ceux-ci sont répartis entre plusieurs groupes (deux au minimum) et reçoivent l’un des médicaments (ou traitements) évalués. A l’issue de l’essai, il est ainsi possible de savoir si le nouveau médicament (ou traitement) fait mieux, aussi bien ou moins bien que le médicament (ou traitement) de référence. C’est essentiellement sur la base des essais de phase III que les médicaments obtiennent (ou pas) une autorisation de mise sur le marché (AMM) et sont commercialisés.
- Les essais de phase IV. Ce type d’essais est réalisé une fois qu’un médicament a obtenu son AMM et est commercialisé. Les essais de phase IV visent à évaluer le médicament dans la « vraie vie », c’est-à-dire auprès de l’ensemble des malades qui le prennent et non pas uniquement sur les malades sélectionnés comme dans les essais de phase I à III. Ces essais permettent notamment de recueillir des informations sur les effets indésirables en précisant la fréquence de leur survenue et en identifiant des effets indésirables peu fréquents qui n’avaient pas été observés jusqu’alors. Ils sont généralement non comparatifs et incluent souvent un grand nombre de malades (plusieurs centaines à plusieurs milliers).
En cancérologie, on assiste ces dernières années à une accélération des processus de recherche clinique. Il est ainsi plutôt fréquent que des essais cliniques soient dits de phase I/II ou II/III. Dans ce cas, il s’agit d’essais qui combinent les objectifs de deux phases.
La méthodologie des essais
Un essai clinique doit apporter une réponse précise à la question posée de la manière la plus rigoureuse possible. La méthodologie est l’ensemble des procédures nécessaires pour obtenir les données qui permettront d’apporter cette réponse. Elles sont consignées dans le protocole de recherche. Cette méthodologie est adaptée en fonction de :
- La phase de l’essai. Les procédures ne sont pas les mêmes selon, par exemple, qu’il s’agit d’un essai de phase I (première administration à l’homme) et de phase III (comparaison avec un traitement de référence).
- La question posée. Un essai qui compare, par exemple, l’efficacité de deux médicaments n’est pas réalisée de la même manière qu’un essai qui cherche à identifier entre deux marqueurs biologiques lequel est le plus pertinent pour suivre l’évolution d’une maladie.
- La maladie concernée. Les procédures tiennent compte des caractéristiques propres à chaque maladie. Par exemple, la durée des traitements n’est pas la même si l’essai concerne une infection aiguë ou une maladie chronique. De même, selon le stade d’une même pathologie, la méthodologie pourra être différente.
Ce sont le promoteur et l’investigateur principal qui déterminent la méthodologie de l’essai lors de la phase de conception de ce dernier et lors de la rédaction du protocole de recherche.
Les différentes étapes de réalisation des essais
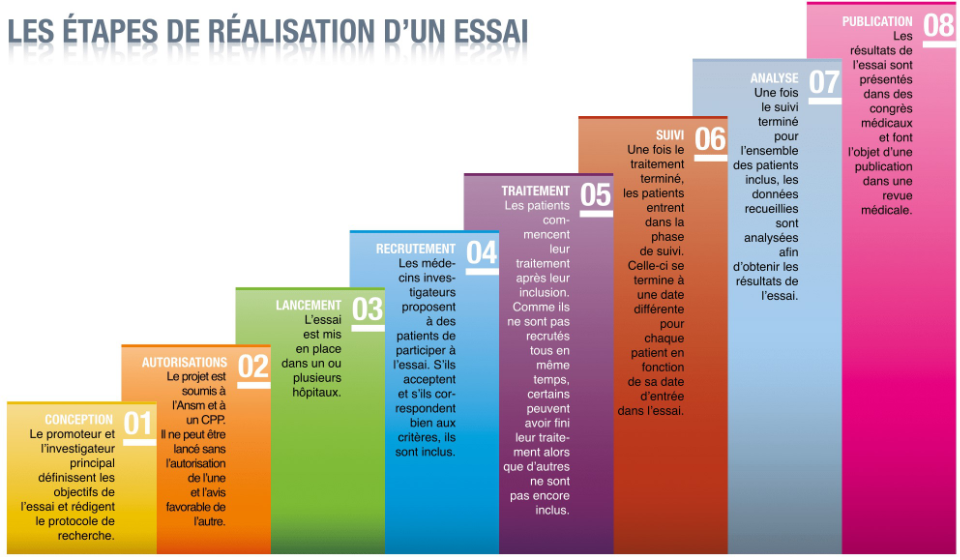
La réalisation des essais cliniques comporte plusieurs étapes :
- La conception : c’est le moment où le promoteur et l’investigateur principal définissent les objectifs de l’essai et rédigent le protocole.
- Les autorisations : le promoteur adresse le protocole à l’ANSM et à un CPP afin d’obtenir l’autorisation de la première et l’avis favorable du second.
- Le lancement de l’essai : le promoteur contacte des centres hospitaliers et des médecins susceptibles de participer à l’essai. En cas d’accord, la logistique nécessaire à la réalisation de l’essai est mise en place dans chacun des centres investigateurs.
- L’inclusion : c’est la période pendant laquelle chacun des médecins investigateurs sélectionne des patients parmi ceux qu’ils suivent et leur proposent d’entrer dans l’essai.
- La phase de traitement : durant cette phase, les patients inclus reçoivent le traitement prévu par le protocole.
- La phase de suivi : une fois le traitement prévu terminé, les patients sont généralement suivis pendant une période plus ou moins longue.
- L’analyse : l’ensemble des données recueillies pendant les phases de traitement et de suivi est analysé afin d’apporter la réponse à la question posée par l’essai.
- La publication : les résultats de l’essai font l’objet d’une publication dans une revue médicale afin d’être partagée avec l’ensemble de la communauté médicale.
Les conditions pour participer à un essai
Pour qu’une réponse à la question posée par un essai puisse être obtenue, il est nécessaire de sélectionner les malades qui vont y participer. Les participants doivent généralement avoir la même maladie, à un stade d’évolution proche et avoir besoin, contre leur maladie, du même type de prise en charge médicale. Le protocole de recherche défini ainsi un certain nombre de critères auxquels doivent répondre les patients. Il y a :
- Les critères d’inclusion. Ce sont les critères nécessaires pour entrer dans l’essai. Ces critères portent en règle générale sur les paramètres de la maladie (stade d’évolution, localisation, traitement antérieur, etc.), des paramètres de santé globale (fonctions rénale et hépatique, nombre des cellules sanguines, etc.), et l’âge (il faut par exemple avoir au moins 18 ans pour participer à un essai clinique réservé aux adultes ; certains essais peuvent s’adresser à des malades d’une catégorie d’âge précise). Il est également nécessaire d’être affilié à un régime d’assurance maladie pour participer à un essai clinique.
- Les critères d’exclusion. Ce sont les critères qui ne permettent pas à un malade de participer à un essai en particulier. Ces critères peuvent concerner la maladie (avoir une maladie étendue alors que l’essai concerne uniquement des malades avec une maladie localisée), d’autres maladies (avoir une atteinte des reins en plus d’un lymphome par exemple), les traitements antérieurement reçus (un essai peut concerner uniquement des malades qui n’ont jamais été traités auparavant), des paramètres de santé globale (avoir des reins ou un cœur qui est altéré par exemple si cela est contre-indiqué avec le ou les traitements évalués).
En fonction de ces différents critères, chaque médecin investigateur d’un essai détermine parmi les patients qu’il suit lesquels sont susceptibles de pouvoir y participer. A l’occasion d’une consultation, il leur apporte une information complète, orale et écrite, et leur fait la proposition d’être inclus dans l’essai.
Les spécificités des essais cliniques en cancérologie
Les essais cliniques concernant des cancers sont réalisés selon les mêmes règles éthiques et méthodologiques que pour toutes les maladies. Ils présentent toutefois certaines spécificités :
- L’absence de volontaires sains. La plupart du temps, les essais de phase I (première administration d’un médicament à l’homme) sont réalisés chez des volontaires sains, c’est-à-dire des personnes en bonne santé. En cancérologie, il n’y a pas de volontaires sains dans les essais, ils sont toujours réalisés avec la participation de personnes malades. D’une part, cela permet de proposer des traitements potentiellement innovants à des patients et de les tester dans les conditions spécifiques de la maladie. D’autre part, les médicaments utilisés en cancérologie exposent souvent à des effets indésirables qui sont parfois importants. Il est donc préférable d’éviter de les administrer à des personnes en bonne santé.
- Le recours limité au placebo. Un placebo est une substance inactive qui se présente comme un vrai médicament. Dans un essai clinique, le fait de donner un placebo à une partie des patients participant permet d’avoir une comparaison très sûre de l’effet du traitement administré à l’autre partie des patients. Cependant, il peut être fait recours au placebo dans un essai essentiellement dans trois types de circonstances :
- Lorsque la maladie concernée est peu grave ;
- Lorsque l’influence de l’état psychologique des patients peut être importante (pour un traitement contre les migraines par exemple)
- Lorsqu’il n’existe aucun traitement de référence contre la maladie concernée.
Ces trois conditions n’existent pas en cancérologie. Il serait même totalement contraire à l’éthique médicale de donner un placebo à des malades atteints d’un cancer. C’est pourquoi il n’existe pas d’essais cliniques en cancérologie où des malades reçoivent un traitement et d’autres un placebo. L’usage du placebo existe toutefois dans certains essais. Dans ce cas, tous les malades reçoivent le traitement de référence qui est associé soit à un nouveau médicament, soit à un placebo. Les patients ne sont alors pas pénalisés parce qu’ils sont tous traités.
Qu’est-ce que la randomisation ?
La randomisation est une procédure qui concerne essentiellement les essais de phase III, qui comparent plusieurs médicaments, traitements ou mode de prise en charge. Elle consiste à attribuer au hasard le médicament ou traitement que va recevoir chacun des patients participant à l’essai. L’équipe en charge de l’essai procède pour cela à un tirage au sort effectué par un ordinateur.
La randomisation permet d’assurer une répartition équitable des traitements de l’essai entre les participants, sans que des facteurs humains entrent en ligne de compte. C’est une procédure essentiellement pour assurer la rigueur scientifique des résultats de l’essai.
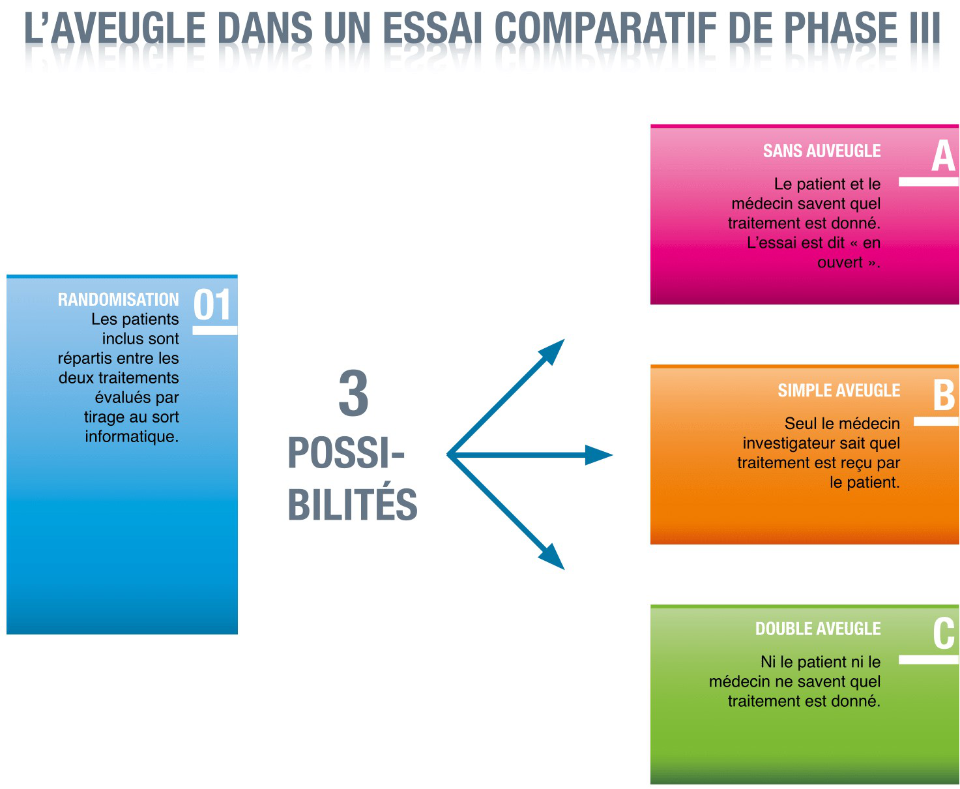
Qu’est-ce que l’aveugle ?
L’aveugle consiste à ce que le traitement reçu par un patient inclus dans un essai ne soit pas connu par celui-ci. Lorsque ni le patient, ni le médecin investigateur n’ont connaissance du traitement attribué, on parle de double aveugle. Il s’agit là encore de s’assurer qu’aucun facteur humain n’entre en ligne de compte dans les résultats de l’essai lorsque celui-ci est comparatif. Si le patient et le médecin savent quel traitement le premier reçoit, cela peut influencer la façon d’évaluer le traitement. Par exemple, le patient qui reçoit le traitement standard et qui aurait préféré recevoir le nouveau traitement peut avoir, de bonne foi, le sentiment qu’il a beaucoup d’effets indésirables. De même, le médecin peut avoir tendance, si son patient reçoit le médicament expérimental d’un essai et s’il croit beaucoup dans ce médicament, à apprécier de façon plus positive son efficacité et à minimiser un peu ses effets indésirables. Le double aveugle permet ainsi de préserver l’objectivité des résultats. Il concerne essentiellement les essais de phase III.